La prevención de la pérdida urinaria excesiva de sodio constituye uno de los pilares fisiológicos más críticos para la estabilidad del volumen extracelular y del volumen plasmático, porque el sodio es el principal osmólito del compartimento extracelular y el determinante primario de su contenido de agua. La evidencia experimental en fisiología renal ha demostrado de manera consistente que las variaciones en el contenido corporal de sodio se traducen en cambios proporcionales del volumen extracelular, debido a que el agua se distribuye osmóticamente siguiendo al sodio en el espacio extracelular, tal como fue establecido en los estudios integradores de balance de sodio y agua en mamíferos realizados por Guyton y colaboradores, quienes demostraron que el control a largo plazo de la presión arterial depende directamente del control renal del sodio corporal total en lugar de depender únicamente del control del tono vascular.
Desde un punto de vista funcional, la razón por la cual la pérdida urinaria excesiva de sodio resulta potencialmente crítica radica en que el organismo humano carece de mecanismos extrarrenales eficaces para compensar pérdidas sostenidas de sodio. Investigaciones fisiológicas clásicas sobre homeostasis del sodio han mostrado que la ingesta dietética y la excreción renal constituyen los dos únicos determinantes relevantes del balance corporal de este catión, de modo que cualquier incremento sostenido en su excreción renal conduce inevitablemente a una contracción del volumen extracelular, como fue descrito en los modelos de equilibrio de sodio desarrollados por Borst y Borst de Geus en sus estudios sobre regulación del volumen extracelular en humanos.
El sodio, además de su papel osmótico, es fundamental para la excitabilidad eléctrica celular, ya que establece el gradiente electroquímico necesario para la generación de potenciales de acción y para el transporte secundario de múltiples solutos. Hille demostró en su análisis de los canales iónicos que el gradiente de sodio a través de la membrana plasmática es la principal fuente de energía electroquímica utilizada por cotransportadores para la entrada de glucosa, aminoácidos y otros nutrientes esenciales, lo que implica que la depleción de sodio extracelular compromete indirectamente la homeostasis metabólica celular.
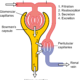
En el riñón, la reabsorción de sodio filtrado se distribuye de manera segmentaria a lo largo de la nefrona, lo que permite una regulación altamente especializada del balance de este ion. Estudios de micropunción y perfusión tubular han demostrado que aproximadamente dos tercios del sodio filtrado se reabsorben en el túbulo proximal, principalmente mediante cotransporte con glucosa, aminoácidos y bicarbonato, así como por intercambio sodio-hidrógeno, como fue caracterizado por Giebisch y Windhager en sus investigaciones sobre transporte tubular proximal en riñón de mamífero. Este segmento actúa como un sistema de reabsorción masiva y poco regulable finamente, cuya función principal es evitar pérdidas excesivas iniciales de sodio filtrado.
El asa de Henle, especialmente su rama ascendente gruesa, desempeña un papel distinto pero complementario, ya que genera el gradiente osmótico corticomedular mediante la reabsorción activa de sodio, potasio y cloro a través del cotransportador sodio-potasio-dos-cloruros. Este mecanismo ha sido descrito por Hebert y Andreoli como el fundamento del mecanismo de concentración urinaria, en el cual la reabsorción de sodio sin acompañamiento de agua permite la creación de un intersticio hiperosmótico que posteriormente facilita la reabsorción de agua en segmentos más distales.
Sin embargo, la regulación fina del contenido final de sodio en la orina ocurre principalmente en el túbulo distal tardío y en los conductos colectores corticales, donde el transporte de sodio se ajusta en tiempo real en función de señales hormonales y del estado hemodinámico del organismo. Brenner y Rector han descrito estos segmentos como el sitio decisivo de control del balance de sodio, debido a que pequeñas variaciones en su actividad reabsortiva producen grandes cambios en la excreción final de sodio, lo que confiere al riñón una capacidad de ajuste altamente sensible del volumen extracelular.
La aldosterona es la hormona más importante en la regulación de este proceso. Investigaciones de Fuller y Young han demostrado que la aldosterona, producida en la zona glomerulosa de la corteza suprarrenal, es estimulada por la activación del sistema renina angiotensina y por incrementos en la concentración plasmática de potasio. A nivel celular, la aldosterona actúa sobre las células principales del conducto colector aumentando la expresión y la actividad de los canales epiteliales de sodio y de la bomba sodio-potasio trifosfatasa de adenosina, lo que incrementa la reabsorción de sodio desde la luz tubular hacia el intersticio. Este mecanismo no solo reduce la excreción de sodio, sino que también favorece la retención de agua de manera indirecta al aumentar la osmolaridad intersticial y al expandir el volumen extracelular.
La importancia fisiológica de este sistema se evidencia en estudios de escape de aldosterona, donde se ha observado que incluso en presencia de niveles elevados de aldosterona, el organismo puede limitar la retención excesiva de sodio mediante mecanismos compensatorios renales, como fue descrito por Stokes en sus estudios sobre adaptación renal a la sobrecarga mineralocorticoide. Esto demuestra que el control del sodio no depende de una única hormona, sino de una red integrada de señales.
La angiotensina II representa otro regulador fundamental del transporte de sodio. Hall y Guyton demostraron en sus estudios sobre el sistema renina angiotensina que la angiotensina II incrementa la reabsorción de sodio en múltiples segmentos de la nefrona, incluyendo el túbulo proximal y el conducto colector, mediante la activación del intercambiador sodio-hidrógeno y la estimulación directa de la actividad del cotransporte tubular. Además, la angiotensina II induce vasoconstricción de la arteriola eferente, lo que aumenta la fracción de filtración y favorece indirectamente la reabsorción proximal de sodio por incremento de las fuerzas de Starling peritubulares.
Las prostaglandinas renales actúan como moduladores contrarreguladores de este sistema. Investigaciones de Harris y Breyer han demostrado que las prostaglandinas, particularmente la prostaglandina E dos, se sintetizan en el riñón en respuesta a estímulos como la angiotensina II y el estrés hemodinámico, y ejercen un efecto vasodilatador sobre la microcirculación renal, además de inhibir la reabsorción de sodio en el túbulo colector. Este efecto permite limitar la retención excesiva de sodio y protege al riñón de una vasoconstricción prolongada, manteniendo el equilibrio entre conservación y excreción.
En conjunto, la evidencia científica demuestra que la prevención de la pérdida urinaria excesiva de sodio no es un proceso pasivo, sino el resultado de una red altamente integrada de transporte tubular segmentario y regulación neurohormonal. Este sistema asegura que el volumen extracelular y el volumen plasmático permanezcan dentro de rangos estrechos compatibles con la perfusión tisular adecuada, la estabilidad hemodinámica y el funcionamiento celular normal, lo que convierte al riñón en el principal órgano de control del balance hidrosalino del organismo.

Fuente y lecturas recomendadas:
- Brenner, B. M., & Rector, F. C. (2008). Brenner & Rector’s The Kidney (8th ed.). Saunders Elsevier.
- Borst, J. G. G., & Borst-de Geus, A. (1963). Hypertension explained by Starling’s theory of circulatory homeostasis. The Lancet, 281(7285), 677–682.
- Fuller, P. J., & Young, M. J. (2005). Mechanisms of mineralocorticoid action. Physiological Reviews, 85(4), 1327–1370.
- Giebisch, G., & Windhager, E. E. (2009). Transport of sodium and chloride in the proximal tubule. Comprehensive Physiology, 1–42.
- Guyton, A. C., Coleman, T. G., & Granger, H. J. (1972). Circulation: overall regulation. Annual Review of Physiology, 34, 13–46.
- Hall, J. E. (1991). The kidney, hypertension, and obesity. Hypertension, 19(1 Suppl), I1–I11.
- Hebert, S. C., & Andreoli, T. E. (1984). Control of NaCl transport in the thick ascending limb of Henle’s loop. American Journal of Physiology, 246(1), F1–F11.
- Hille, B. (2001). Ion Channels of Excitable Membranes (3rd ed.). Sinauer Associates.
- Harris, R. C., & Breyer, M. D. (2001). Physiological regulation of cyclooxygenase-2 in the kidney. American Journal of Physiology, 281(1), F1–F11.
- Stokes, J. B. (1981). Sodium transport in the cortical collecting tubule. American Journal of Physiology, 241(1), F1–F8.
Síguenos en X: @el_homomedicus y @enarm_intensivo Síguenos en instagram: homomedicus y en Treads.net como: Homomedicus
Aprende administración paso a paso

ADMINISTRACION DESDE CERO